DISC conducts research by developing and applying appropriate data science methods for bioinformatics and computational biology research, in collaboration with faculty, staff, and stakeholders within and outside Tufts University. Some of current work focuses on single-cell transcriptomics, biological networks, and proteomics. Below are some details about our current projects.
Jump to a Project
- Single Cell Profiling of Hofbauer Cells and Fetal Brain Microglia Reveals Shared Programs and Functions
- Neutrophils and Macrophages Drive TNF-induced Lethality via TRIF/CD14-mediated Responses
- Multi-resolution Characterization of Molecular Taxonomies in Transcriptomics Data
- System-Level Analysis of Omics Data to Reveal Mechanisms of Head & Neck Cancer
- Proteomics Profiling Approaches to Study Healthy Aging and Longevity
Single Cell Profiling of Hofbauer Cells and Fetal Brain Microglia Reveals Shared Programs and Functions
Collaboration between Tufts DISC (Rebecca Batorsky), Tufts University Computer Science, Duke University Psychology and Neuroscience and Division of Maternal-Fetal Medicine and Lurie Center for Autism Massachusetts General Hospital
Maternal immune activation is associated with adverse offspring neurodevelopmental outcomes, many of which are mediated by in utero microglial programming. Microglia remain inaccessible at birth and throughout development, thus identification of noninvasive biomarkers that can reflect fetal brain microglial programming may permit screening and intervention during critical developmental windows. Here we used lineage tracing to demonstrate the shared ontogeny between fetal brain macrophages (microglia) and fetal placental macrophages (Hofbauer cells). Single-cell RNA sequencing of murine fetal brain and placental macrophages demonstrated shared transcriptional programs.
For more information contact Rebecca Batorky
References:
- Ceasrine, A.M., Batorsky, R., Shook, L.L., Kislal, S., Bordt, E.A., Devlin, B.A., Perlis, R.H., Slonim, D.K., Bilbo, S.D. and Edlow, A.G., 2021. Single cell profiling of Hofbauer cells and fetal brain microglia reveals shared programs and functions. bioRxiv 2021.12.03.471177 [View PDF]
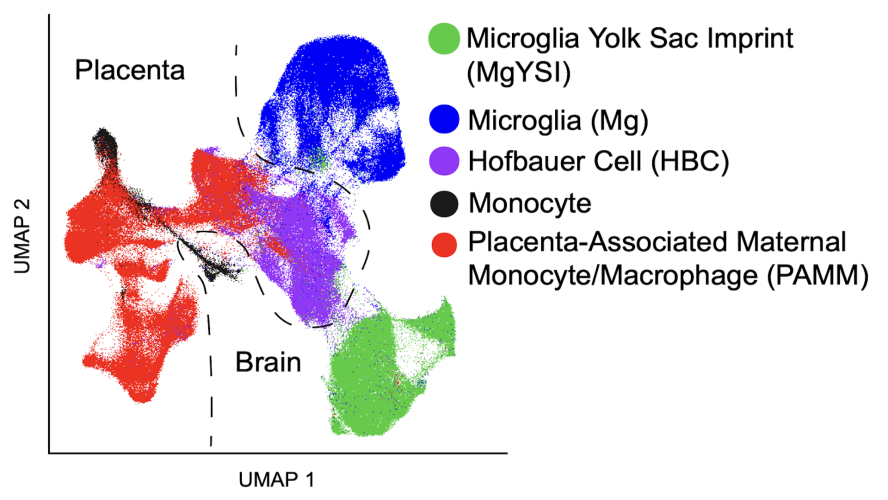 Uniform Manifold Approximation and Projection (UMAP) visualization of microglia/monocyte cells reveals 5 distinct clusters
Uniform Manifold Approximation and Projection (UMAP) visualization of microglia/monocyte cells reveals 5 distinct clusters
Neutrophils and Macrophages Drive TNF-induced Lethality via TRIF/CD14-mediated Responses
Collaboration between Tufts DISC (Rebecca Batorsky), Tufts Department of Immunology, Tufts University School of Medicine and the Department of Infectious Diseases and Global Health, Tufts University Cummings School of Veterinary Medicine
TNF mediates a variety of biological processes including cellular proliferation, inflammatory responses, and cell death and is therefore associated with numerous pathologies including autoinflammatory diseases and septic shock. The inflammatory and cell death responses to TNF have been studied extensively downstream of TNF-R1 and are believed to rely on the formation of proinflammatory complex I and prodeath complex II, respectively. We recently identified a similar multimeric complex downstream of TLR4, termed the TRIFosome, that regulates inflammation and cell death in response to LPS or Yersinia pseudotuberculosis. We present evidence of a role for the TRIFosome downstream of TNF-R1, independent of TLR3 or TLR4 engagement. Specifically, TNF-induced cell death and inflammation in murine macrophages were driven by the TLR4 adaptor TRIF and the LPS co-receptor CD14, highlighting an important role for these proteins beyond TLR-mediated immune responses. Via immunoprecipitation and visualization of TRIF-specific puncta, we demonstrated TRIF- and CD14-dependent formation of prodeath and proinflammatory complexes in response to TNF. Extending these findings, in a murine TNF-induced sepsis model, TRIF and CD14 deficiency decreased systemic inflammation, reduced organ pathology, and improved survival. Single-cell RNA sequencing demonstrated that the outcome of TRIF activation was cell specific, because TNF-induced lethality was mediated by neutrophils and macrophages responding to TNF in a TRIF-dependent manner. Our findings suggest that in addition to their crucial role in TNF production, myeloid cells are central to TNF toxicity and position TRIF and CD14 as universal components of receptor-mediated immune responses.
For more information contact Rebecca Batorky
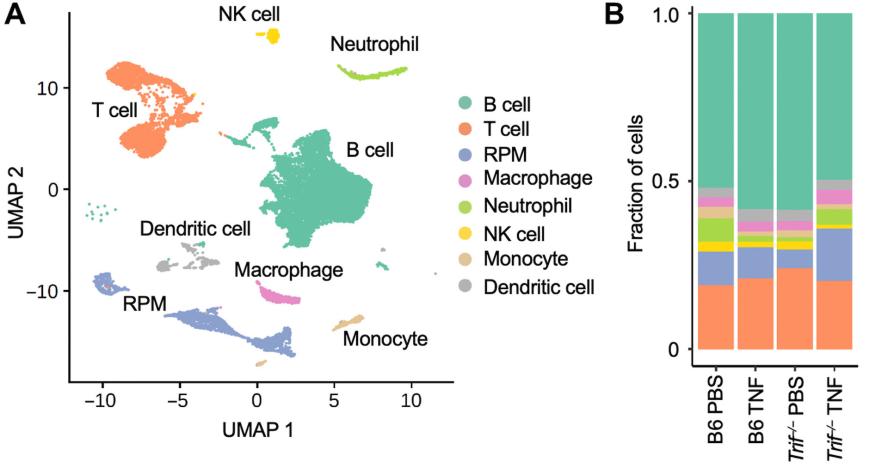 Uniform Manifold Approximation and Projection analysis of splenocytes from mice reveals 8 distinct clusters. The right plot shows the fraction of cells corresponding to each cell type.
Uniform Manifold Approximation and Projection analysis of splenocytes from mice reveals 8 distinct clusters. The right plot shows the fraction of cells corresponding to each cell type.
References:
- H. I. Muendlein, W. M. Connolly, J. Cameron, D. Jetton, Z. Magri, I. Smirnova, E. Vannier, X. Li, R.E. Batorsky, A. Poltorak., 2023. “Neutrophils and macrophages drive TNF-induced lethality via TRIF/CD14-mediated responses,” Science Immunology, Vol. 7, No. 78. [View PDF]
Multi-resolution Characterization of Molecular Taxonomies in Transcriptomics Data
Collaboration between Tufts DISC (Eric Reed), BERD and QMDS Centers of Tufts University School of Medicine, and the Boston University Center for Computational Biomedicine at Boston University School of Medicine
“Omics” studies are often explorative, in that relationships between samples are not explicitly defined a priori, but rather emerge from data-driven discovery and annotation of molecular subtypes, thereby informing hypotheses and independent evaluation. Moreover, “omics” studies give way to various data paradigms, calling for adaptable approaches to generate useful models. To this end, we developed K2Taxonomer, an unsupervised recursive partitioning algorithm and associated R package that utilize ensemble learning to identify robust subgroups in a 'taxonomy-like' structure, and is suitable for the analysis of both bulk and single-cell transcriptomics, and other '-omics' data. Accordingly, this method has been applied to in a multitude of projects spanning numerous biomedical fields, including: toxicogenomics, breast cancer, head and neck cancer, and aging.
For more information contact Eric Reed
References:
- Karagiannis, T., Dowrey, T., Villacorta-Martin, C., Montano, M., Reed, E., Andersen, S., Perls, T., Monti, S., Murphy, G. and Sebastiani, P., 2022. "Multi-modal profiling of peripheral blood cells across the human lifespan reveals distinct immune cell signatures of aging and longevity." Submitted to Immunology. [View PDF]
- Reed, E.R. and Monti, S., 2021. Multi-resolution characterization of molecular taxonomies in bulk and single-cell transcriptomics data. Nucleic acids research, 49(17), pp.e98-e98. [View PDF]
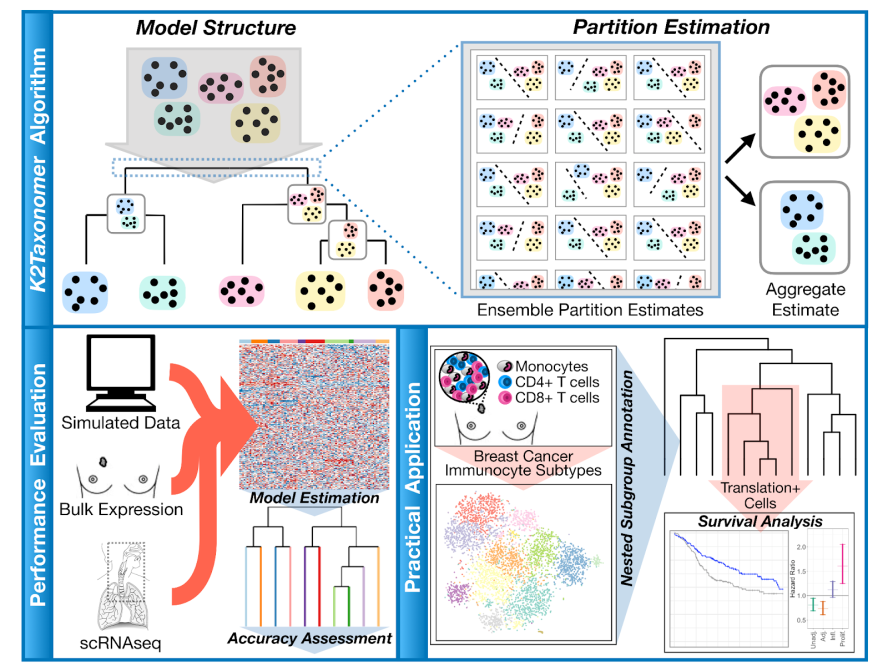 K2Taxonomer: an unsupervised recursive partitioning algorithm identifies robust subgroups in a 'taxonomy-like' structure
K2Taxonomer: an unsupervised recursive partitioning algorithm identifies robust subgroups in a 'taxonomy-like' structure
3. Kim, S., Reed, E., Monti, S. and Schlezinger, J.J., 2021. “A data-driven transcriptional taxonomy of adipogenic chemicals to identify white and brite adipogens,” Environmental health perspectives, 129(7), p.077006. [View PDF]
System-Level Analysis of Omics Data to Reveal Mechanisms of Head & Neck Cancer
Collaboration between Tufts DISC (Eric Reed) and the Boston University Center for Computational Biomedicine at Boston University School of Medicine
To support the design of more effective targeted treatments, our project focuses on better understanding of the regulatory mechanisms governing the maintenance, expansion and evolution of these cell populations. From an ‘omics’ analysis perspective, this means moving beyond standard one-gene-at-a-time types of analysis whereby differential analyses are performed to rank genes and pathways based on their differential behavior between phenotypic groups. Accordingly, this project focuses on the refinement of system-level network-based approaches and development of methods to compare networks to elucidate the molecular rewiring (i.e., changes in gene connectivity) occurring in response to perturbation (e.g., treatment) or between distinct phenotypic groups. This approach will not only identify differential markers, but will also point to important hubs, their cross-talk, and the targets they regulate, and suggest relevant manipulations.
For more information contact Eric Reed
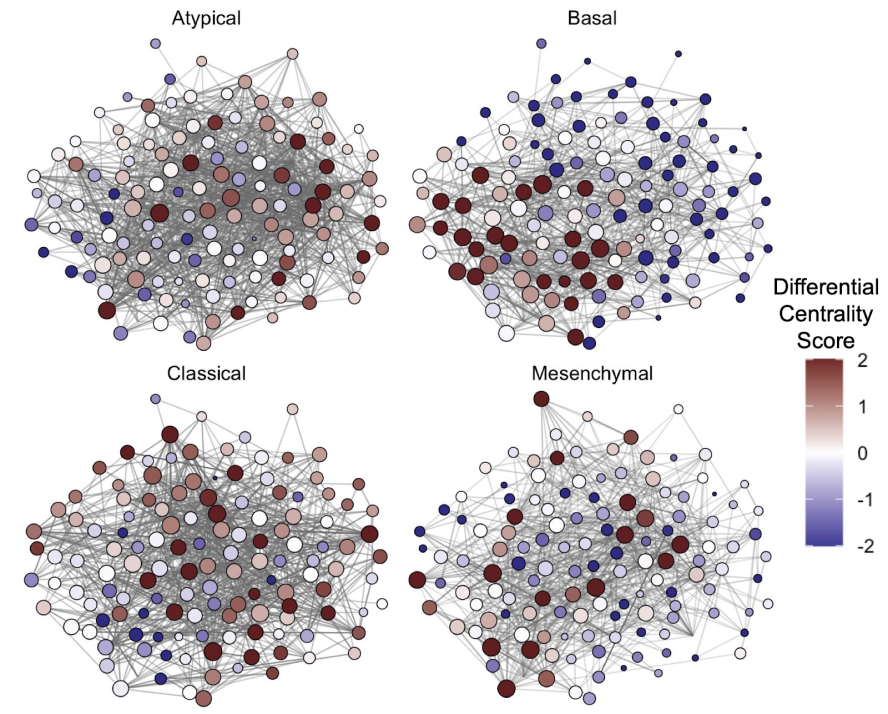 Biological networks and differential centrality score for 4 different types of cells in head and neck cancer
Biological networks and differential centrality score for 4 different types of cells in head and neck cancer
Proteomics Profiling Approaches to Study Healthy Aging and Longevity
Collaboration between Tufts DISC (Eric Reed) and the BERD and QMDS Centers of Tufts University School of Medicine
The emerging practicality of proteomics profiling is particularly exciting given that, unlike their coding RNA transcripts, proteins are primary effectors of biological systems. In the space of aging and age-related diseases, numerous studies have reported dynamic proteomic changes and numerous protein biomarkers have been reported. However, the performance of emerging high-throughput proteomics platforms has yet to be adequately assessed, such that the reproducibility of these proteomics signatures remains uncertain. Accordingly, the primary focus of this project is comparative evaluation of three proteomics technologies, including both “probe-based” methods and mass spectrometry. To this end, we are working to evaluate the sensitivity and specificity of these platforms to detect varying concentrations of proteins spiked into human serum samples. In a related project, we are developing statistical approaches to identify technically conserved protein signatures of extreme old-age based on human plasma profiles of the same subjects generated by multiple platforms.
For more information contact Eric Reed
 Grid display of serum proteins for which association of measured expression with extreme old age is conserved across multiple proteomics. Collectively, these results signify high-confidence biological drivers of aging and healthy aging
Grid display of serum proteins for which association of measured expression with extreme old age is conserved across multiple proteomics. Collectively, these results signify high-confidence biological drivers of aging and healthy aging